Configuring INI-files: Examples
This page provides the Quick start guidelines and examples of the config.ini files for six common scenarios using (a combination of) the following steps:
Demultiplexing (Cutadapt).
Trimming: Trimming barcodes, spacers, and restriction enzyme cutsite remnants at the 5’ end of the reads (while compensating for variable length barcodes and spacers by trimming at the 3’ end of forward reads) and trimming of restriction enzyme cutsite remnants, barcodes, and adapter sequences at the 3’ end of the reads. (Cutadapt).
Merging of forward and reverse reads.
Removal of reads with low quality base-calling (Python).
Removal of reads with internal restriction sites (Python).
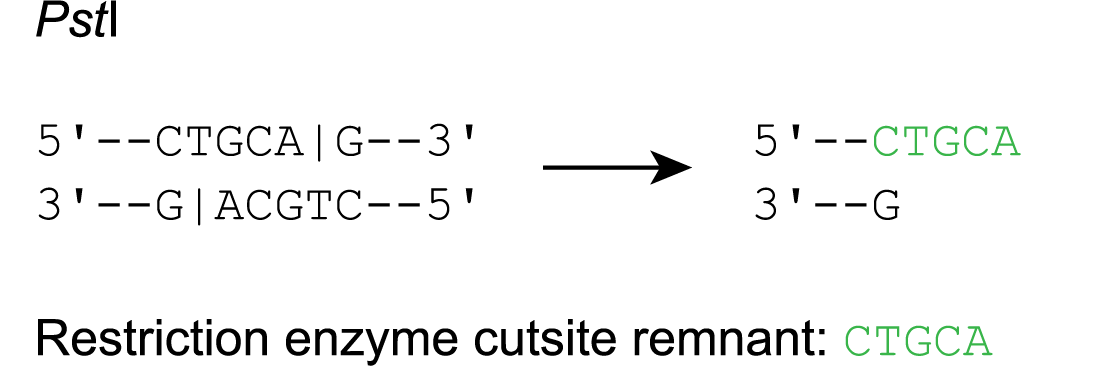
Restriction enzyme cutsite remnants and sequencing primers
- An overhang remains on the top strand (e.g. Pst I)
In this case, the RE cutsite remnant is equal to the top strand overhang.
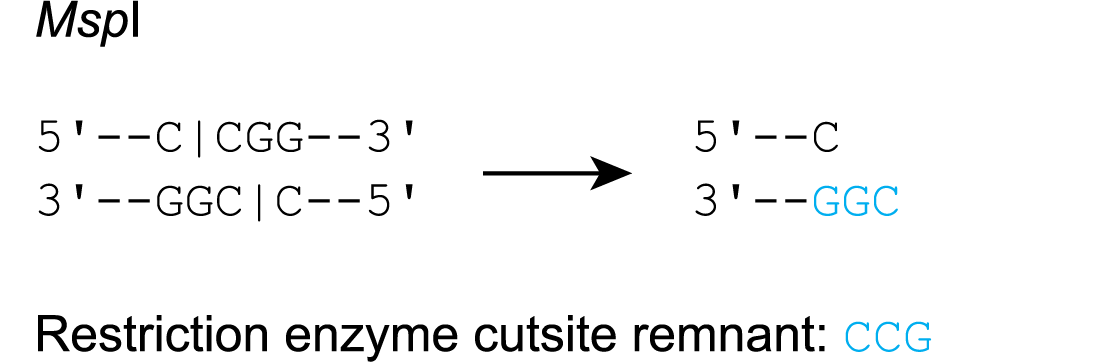
- An overhang remains on the bottom strand (e.g. Msp I)
In this case, the RE cutsite remnant is equal to the complement of the bottom strand overhang.
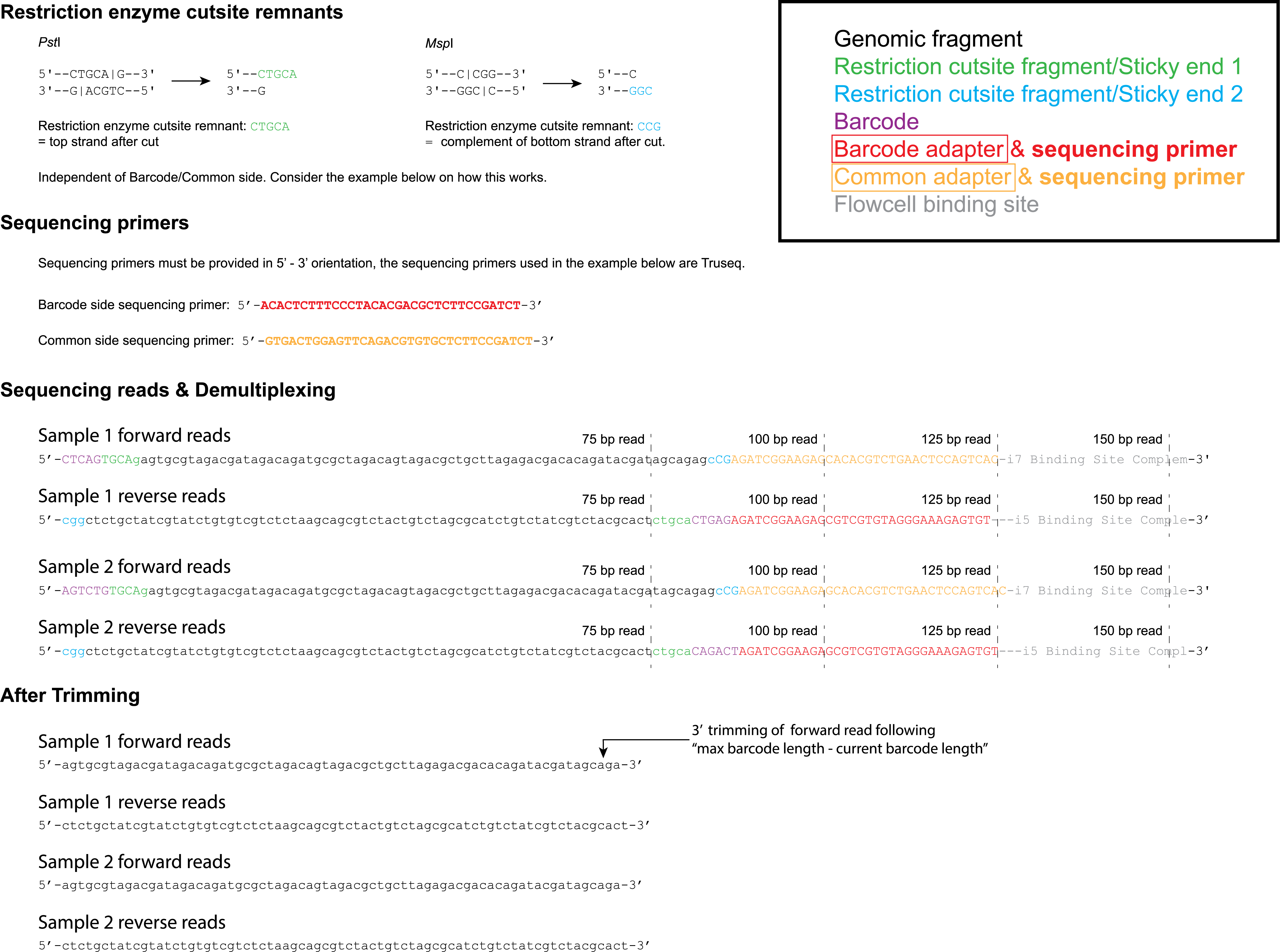
For both scenario’s the RE cutsite remnant is independent of the side (barcode or common). Consider the elaborate library preparation example of a double-digest paired-end sequencing locus in the third tab for further understanding.
Currently two pairs of Illumina sequencing primers are commercially available; Nextera and Truseq. It is important to provide these sequences in 5’ 3’ orientation. Alternatively one can simply fill in the values “Nextera” or “Truseq” of which the corresponding sequences are saved in the algorithm’s memory. The sequences shown below are the Truseq sequencing primers. For further understanding on how these primers are incorporated in the library construct/sequencing result, consider the example in the third tab.


Examples starting with Demultiplexing
Single-digest GBS and single-end sequencing

[General]
cores = 32
input_directory = /home/User/GBS_preprocessing
sequencing_type = se
temp_dir = /tmp/
input_file_name_template = {run:24}_R{orientation:1}_001{extension:10}
# Example: 17146FL-13-01-01_S9_L002_R1_001.fastq.bz2 => run = 17146FL-13-01-01_S9_L002; orientation = 1; extension = .fastq.bz2
# Essential information on run, read orientation, and file extension is obtained from the structure of the name of the original fastq file as provided by the service provider.
# {:XX} mark the number of characters in the file name that contain the specified information. One wildcard is allowed, this is invoked by leaving the character length information out. E.g. {run}_R{orientation:1}{extension:10} .
# Fields "orientation" and "extension" are automatically transferred to all new file names created in the next steps.
[CutadaptDemultiplex]
barcodes = /home/User/GBS_preprocessing/barcodes.fasta
error_rate = 0
output_directory = ./01_demultiplex
output_file_name_template = {sample_name}_{orientation}{extension}
anchored_barcodes=True
barcode_side_cutsite_remnant = CTGCA
# the (barcode + barcode_side_cutsite_remnant) sequence is searched per read, to increase specificity of demultiplexing.
# cutsite remnants may also be provided in separate .fasta files where each sample is followed by their corresponding cutsite remnant.
# Error rate is expressed as fraction of the searched length (range 0.0 - 1.0). 0 means exact match is required. If e.g. barcodes used are 5 bp long and the barcode side RE is PstI (5bp), and a maximum of 1 mismatch is desired, then error_rate should be 0.1.
[CutadaptTrimmer]
barcodes = /home/User/GBS_preprocessing/barcodes.fasta
barcode_side_cutsite_remnant = CTGCA
common_side_cutsite_remnant = CTGCA
# cutsite remnants may also be provided in separate .fasta files where each sample is followed by their corresponding cutsite remnant.
common_side_sequencing_primer = TruSeq
# common_side_sequencing_primer = Nextera
# common_side_sequencing_primer = GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT : TruSeq Illumina sequencing primer on the common (i7) side (5' - 3').
# common_side_sequencing_primer = CGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT : Nextera Illumina sequencing primer on the common (i7) side (5' - 3').
minimum_length = 30
error_rate = 0
output_directory = ./02_trimming
output_file_name_template = {sample_name}_{orientation}{extension}
# Single-End sequencing always starts from the barcode adapter side (i5).
# Although the barcode sequencing primer is the only primer used, the barcode side adapter is never present in the sequencing reads and should therefore not be provided.
# In single-digest GBS, the restriction enzyme cutsite remnant at the common adapter side is the same as on the barcode adapter side.
# Trimming starts by removing the barcode and restriction enzyme cutside remnant at the start of the 5' of the read, and additionally it 3' trims the difference between the maximum barcode length and the current sample's barcode length for each individual read.
# Then it searches for the reverse complement sequence of the common adapter fused to the restriction enzyme cutside remnant and trims these from the 3' end of the read.
# Error rate is expressed as fraction of the searched length (range 0.0 - 1.0). 0 means exact match is required. If e.g. for forward reads Truseq adapters (34bp) and MseI (3bp) as a common side RE were used, and a maximum of 1 mismatch is desired, then the error rate should be 0.028. Note that this only allows 1 mismatch if the entire sequence is found. If a smaller portion of the sequence is found, no mismatches will be allowed.
[MaxNFilter]
max_n = 0
output_directory = ./03_max_n_filter
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with N base calls.
[SlidingWindowQualityFilter]
window_size = 2
average_quality = 20
count = 1
output_directory = ./04_sliding_window
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with low quality base calls.
# Translation of default values: For any given read, if any 2 consecutive bases (window_size) have an average Phred quality lower than 20 (average_quality) at least 1 (count) time, then remove the read.
[AverageQualityFilter]
average_quality = 25
output_directory = ./05_average_quality_filter
output_file_name_template = {sample_name}_{run}{extension}
# removes low quality reads.
[RemovePatternFilter]
pattern = CTGCAG
output_directory = ./06_remove_chimera_partial_digest
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with intact internal restriction enzyme recognition sites.
Single-digest GBS and paired-end sequencing

[General]
cores = 32
input_directory = /home/User/GBS_preprocessing
sequencing_type = pe
temp_dir = /tmp/
input_file_name_template = {run:24}_R{orientation:1}_001{extension:10}
# Example: 17146FL-13-01-01_S9_L002_R1_001.fastq.bz2 => run = 17146FL-13-01-01_S9_L002; orientation = 1; extension = .fastq.bz2
# Essential information on run, read orientation, and file extension is obtained from the structure of the name of the original fastq file as provided by the service provider.
# {:XX} mark the number of characters in the file name that contain the specified information. One wildcard is allowed, this is invoked by leaving the character length information out. E.g. {run}_R{orientation:1}{extension:10} .
# Fields "orientation" and "extension" are automatically transferred to all new file names created in the next steps.
[CutadaptDemultiplex]
barcodes = /home/User/GBS_preprocessing/barcodes.fasta
error_rate = 0
output_directory = ./01_demultiplex
output_file_name_template = {sample_name}_{orientation}{extension}
anchored_barcodes=True
barcode_side_cutsite_remnant = CTGCA
# the (barcode + barcode_side_cutsite_remnant) sequence is searched per Forward read, to increase specificity of demultiplexing.
# cutsite remnants may also be provided in separate .fasta files where each sample is followed by their corresponding cutsite remnant.
# Error rate is expressed as fraction of the searched length (range 0.0 - 1.0). 0 means exact match is required. If e.g. barcodes used are 5 bp long and the barcode side RE is PstI (5bp), and a maximum of 1 mismatch is desired, then error_rate should be 0.1.
[CutadaptTrimmer]
barcodes = /home/User/GBS_preprocessing/barcodes.fasta
barcode_side_cutsite_remnant = CTGCA
# cutsite remnants may also be provided in separate .fasta files where each sample is followed by their corresponding cutsite remnant.
barcode_side_sequencing_primer = TruSeq
# barcode_side_sequencing_primer = Nextera
# barcode_side_sequencing_primer = ACACTCTTTCCCTACACGACGCTCTTCCGATCT : TruSeq and Nextera Illumina sequencing primer on the barcode (i5) side (5' - 3').
common_side_cutsite_remnant = CTGCA
common_side_sequencing_primer = Truseq
# common_side_sequencing_primer = Nextera
# common_side_sequencing_primer = GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT : TruSeq Illumina sequencing primer on the common (i7) side (5' - 3').
# common_side_sequencing_primer = CGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT : Nextera Illumina sequencing primer on the common (i7) side (5' - 3').
minimum_length = 30
error_rate = 0
output_directory = ./02_trimming
output_file_name_template = {sample_name}_{orientation}{extension}
# Trimming searches and trims the barcode and restriction enzyme cutside remnant at the start of the 5' Forward read, additionally it 3' trims the difference between the maximum barcode length and the current sample's barcode length for each individual read.
# Trimming searches and trims the restriction enzyme cutside remnant at the start of the 5' reverse read.
# Trimming searches for the reverse complement sequence of the common adapter fused to the restriction enzyme cutside remnant and trims these from the 3' of the Forward read.
# Trimming searches for the reverse complement sequence of the barcode adapter fused to the sample-specific barcode and the restriction enzyme cutside remnant and trims these from the 3' of the Reverse read.
# In single-digest GBS, the restriction enzyme cutsite remnant at the common adapter side is the same as on the barcode adapter side.
# Error rate is expressed as fraction of the searched length (range 0.0 - 1.0). 0 means exact match is required. If e.g. for forward reads Truseq adapters (34bp) and MseI (3bp) as a common side RE were used, and a maximum of 1 mismatch is desired, then the error rate should be 0.028. Note that this only allows 1 mismatch if the entire sequence is found. If a smaller portion of the sequence is found, no mismatches will be allowed.
[MaxNFilter]
max_n = 0
output_directory = ./03_max_n_filter
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with N base calls.
[SlidingWindowQualityFilter]
window_size = 2
average_quality = 20
count = 1
output_directory = ./04_sliding_window
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with low quality base calls.
# Translation of default values: For any given read, if any 2 consecutive bases (window_size) have an average Phred quality lower than 20 (average_quality) at least 1 (count) time, then remove the read.
[AverageQualityFilter]
average_quality = 25
output_directory = ./05_average_quality_filter
output_file_name_template = {sample_name}_{run}{extension}
# removes low quality reads.
[RemovePatternFilter]
pattern = CTGCAG
# first enzyme
output_directory = ./06_remove_chimera_partial_digest
output_file_name_template = {sample_name}_{run}{extension}

[General]
cores = 32
input_directory = /home/User/GBS_preprocessing
sequencing_type = pe
temp_dir = /tmp/
input_file_name_template = {run:24}_R{orientation:1}_001{extension:10}
# Example: 17146FL-13-01-01_S9_L002_R1_001.fastq.bz2 => run = 17146FL-13-01-01_S9_L002; orientation = 1; extension = .fastq.bz2
# Essential information on run, read orientation, and file extension is obtained from the structure of the name of the original fastq file as provided by the service provider.
# {:XX} mark the number of characters in the file name that contain the specified information. One wildcard is allowed, this is invoked by leaving the character length information out. E.g. {run}_R{orientation:1}{extension:10} .
# Fields "orientation" and "extension" are automatically transferred to all new file names created in the next steps.
[CutadaptDemultiplex]
barcodes = /home/User/GBS_preprocessing/barcodes.fasta
error_rate = 0
output_directory = ./01_demultiplex
output_file_name_template = {sample_name}_{orientation}{extension}
barcode_side_cutsite_remnant = CTGCA
anchored_barcodes=True
# the (barcode + barcode_side_cutsite_remnant) sequence is searched per Forward read, to increase specificity of demultiplexing.
# cutsite remnants may also be provided in separate .fasta files where each sample is followed by their corresponding cutsite remnant.
# Error rate is expressed as fraction of the searched length (range 0.0 - 1.0). 0 means exact match is required. If e.g. barcodes used are 5 bp long and the barcode side RE is PstI (5bp), and a maximum of 1 mismatch is desired, then error_rate should be 0.1.
[CutadaptTrimmer]
barcodes = /home/User/GBS_preprocessing/barcodes.fasta
barcode_side_cutsite_remnant = CTGCA
# cutsite remnants may also be provided in separate .fasta files where each sample is followed by their corresponding cutsite remnant.
barcode_side_sequencing_primer = TruSeq
# barcode_side_sequencing_primer = Nextera
# barcode_side_sequencing_primer = ACACTCTTTCCCTACACGACGCTCTTCCGATCT : TruSeq and Nextera Illumina sequencing primer on the barcode (i5) side (5' - 3').
common_side_cutsite_remnant = CTGCA
common_side_sequencing_primer = Truseq
# common_side_sequencing_primer = Nextera
# common_side_sequencing_primer = GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT : TruSeq Illumina sequencing primer on the common (i7) side (5' - 3').
# common_side_sequencing_primer = CGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT : Nextera Illumina sequencing primer on the common (i7) side (5' - 3').
minimum_length = 30
error_rate = 0
output_directory = ./02_trimming
output_file_name_template = {sample_name}_{orientation}{extension}
# Trimming searches and trims the barcode and restriction enzyme cutside remnant at the start of the 5' Forward read, additionally it 3' trims the difference between the maximum barcode length and the current sample's barcode length for each individual read.
# Trimming searches and trims the restriction enzyme cutside remnant at the start of the 5' reverse read.
# Trimming searches for the reverse complement sequence of the common adapter fused to the restriction enzyme cutside remnant and trims these from the 3' of the Forward read.
# Trimming searches for the reverse complement sequence of the barcode adapter fused to the sample-specific barcode and the restriction enzyme cutside remnant and trims these from the 3' of the Reverse read.
# In single-digest GBS, the restriction enzyme cutsite remnant at the common adapter side is the same as on the barcode adapter side.
# Error rate is expressed as fraction of the searched length (range 0.0 - 1.0). 0 means exact match is required. If e.g. for forward reads Truseq adapters (34bp) and MseI (3bp) as a common side RE were used, and a maximum of 1 mismatch is desired, then the error rate should be 0.028. Note that this only allows 1 mismatch if the entire sequence is found. If a smaller portion of the sequence is found, no mismatches will be allowed.
[Pear]
minimum_length = 40
minimum_overlap = 10
output_directory = ./03_merging
output_file_name_template = {sample_name}_{run}.assembled{extension}
# merges Forward and Reverse reads by sequence overlap.
[MaxNFilter]
max_n = 0
output_directory = ./04_max_n_filter
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with N base calls.
[SlidingWindowQualityFilter]
window_size = 2
average_quality = 20
count = 1
output_directory = ./05_sliding_window
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with low quality base calls.
# Translation of default values: For any given read, if any 2 consecutive bases (window_size) have an average Phred quality lower than 20 (average_quality) at least 1 (count) time, then remove the read.
[AverageQualityFilter]
average_quality = 25
output_directory = ./06_average_quality_filter
output_file_name_template = {sample_name}_{run}{extension}
# removes low quality reads.
[RemovePatternFilter]
pattern = CTGCAG
# first enzyme
output_directory = ./07_remove_chimera_partial_digest
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with intact internal restriction enzyme recognition sites.
Double-digest GBS and single-end sequencing

[General]
cores = 32
input_directory = /home/User/GBS_preprocessing
sequencing_type = se
temp_dir = /tmp/
input_file_name_template = {run:24}_R{orientation:1}_001{extension:10}
# Example: 17146FL-13-01-01_S9_L002_R1_001.fastq.bz2 => run = 17146FL-13-01-01_S9_L002; orientation = 1; extension = .fastq.bz2
# Essential information on run, read orientation, and file extension is obtained from the structure of the name of the original fastq file as provided by the service provider.
# {:XX} mark the number of characters in the file name that contain the specified information. One wildcard is allowed, this is invoked by leaving the character length information out. E.g. {run}_R{orientation:1}{extension:10} .
# Fields "orientation" and "extension" are automatically transferred to all new file names created in the next steps.
[CutadaptDemultiplex]
barcodes = /home/User/GBS_preprocessing/barcodes.fasta
error_rate = 0
output_directory = ./01_demultiplex
output_file_name_template = {sample_name}_{orientation}{extension}
anchored_barcodes=True
barcode_side_cutsite_remnant = CTGCA
# the (barcode + barcode_side_cutsite_remnant) sequence is searched per read, to increase specificity of demultiplexing.
# cutsite remnants may also be provided in separate .fasta files where each sample is followed by their corresponding cutsite remnant.
# Error rate is expressed as fraction of the searched length (range 0.0 - 1.0). 0 means exact match is required. If e.g. barcodes used are 5 bp long and the barcode side RE is PstI (5bp), and a maximum of 1 mismatch is desired, then error_rate should be 0.1.
[CutadaptTrimmer]
barcodes = /home/User/GBS_preprocessing/barcodes.fasta
barcode_side_cutsite_remnant = CTGCA
common_side_cutsite_remnant = CCG
# cutsite remnants may also be provided in separate .fasta files where each sample is followed by their corresponding cutsite remnant.
common_side_sequencing_primer = TruSeq
# common_side_sequencing_primer = Nextera
# common_side_sequencing_primer = GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT : TruSeq Illumina sequencing primer on the common (i7) side (5' - 3').
# common_side_sequencing_primer = CGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT : Nextera Illumina sequencing primer on the common (i7) side (5' - 3').
minimum_length = 30
error_rate = 0
output_directory = ./02_trimming
output_file_name_template = {sample_name}_{orientation}{extension}
# Single-End sequencing always starts from the barcode adapter side (i5).
# Although the barcode sequencing primer is the only primer used, the barcode side adapter is never present in the sequencing reads and should therefore not be provided.
# Trimming starts by removing the barcode and restriction enzyme cutside remnant at the start of the 5' of the read, and additionally it 3' trims the difference between the maximum barcode length and the current sample's barcode length for each individual read.
# Then it searches for the reverse complement sequence of the common adapter fused to the restriction enzyme cutside remnant and trims these from the 3' end of the read.
# Error rate is expressed as fraction of the searched length (range 0.0 - 1.0). 0 means exact match is required. If e.g. for forward reads Truseq adapters (34bp) and MseI (3bp) as a common side RE were used, and a maximum of 1 mismatch is desired, then the error rate should be 0.028. Note that this only allows 1 mismatch if the entire sequence is found. If a smaller portion of the sequence is found, no mismatches will be allowed.
[MaxNFilter]
max_n = 0
output_directory = ./03_max_n_filter
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with N base calls.
[SlidingWindowQualityFilter]
window_size = 2
average_quality = 20
count = 1
output_directory = ./04_sliding_window
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with low quality base calls.
# Translation of default values: For any given read, if any 2 consecutive bases (window_size) have an average Phred quality lower than 20 (average_quality) at least 1 (count) time, then remove the read.
[AverageQualityFilter]
average_quality = 25
output_directory = ./05_average_quality_filter
output_file_name_template = {sample_name}_{run}{extension}
# removes low quality reads.
[RemovePatternFilter.CTGCAG]
pattern = CTGCAG
# first enzyme
output_directory = ./06_remove_chimera_partial_digest
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with intact internal restriction enzyme recognition sites.
[RemovePatternFilter.CCGG]
pattern = CCGG
# second enzyme
output_directory = ./07_remove_chimera_partial_digest
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with intact internal restriction enzyme recognition sites.
Double-digest GBS and paired-end sequencing
[General]
cores = 32
input_directory = /home/User/GBS_preprocessing
sequencing_type = pe
temp_dir = /tmp/
input_file_name_template = {run:24}_R{orientation:1}_001{extension:10}
# Example: 17146FL-13-01-01_S9_L002_R1_001.fastq.bz2 => run = 17146FL-13-01-01_S9_L002; orientation = 1; extension = .fastq.bz2
# Essential information on run, read orientation, and file extension is obtained from the structure of the name of the original fastq file as provided by the service provider.
# {:XX} mark the number of characters in the file name that contain the specified information. One wildcard is allowed, this is invoked by leaving the character length information out. E.g. {run}_R{orientation:1}{extension:10} .
# Fields "orientation" and "extension" are automatically transferred to all new file names created in the next steps.
[CutadaptDemultiplex]
barcodes = /home/User/GBS_preprocessing/barcodes.fasta
error_rate = 0
output_directory = ./01_demultiplex
output_file_name_template = {sample_name}_{orientation}{extension}
anchored_barcodes=True
barcode_side_cutsite_remnant = CTGCA
# the (barcode + barcode_side_cutsite_remnant) sequence is searched per read, to increase specificity of demultiplexing.
# cutsite remnants may also be provided in separate .fasta files where each sample is followed by their corresponding cutsite remnant.
# Error rate is expressed as fraction of the searched length (range 0.0 - 1.0). 0 means exact match is required. If e.g. barcodes used are 5 bp long and the barcode side RE is PstI (5bp), and a maximum of 1 mismatch is desired, then error_rate should be 0.1.
[CutadaptTrimmer]
barcodes = /home/User/GBS_preprocessing/barcodes.fasta
barcode_side_cutsite_remnant = CTGCA
# cutsite remnants may also be provided in separate .fasta files where each sample is followed by their corresponding cutsite remnant.
barcode_side_sequencing_primer = TruSeq
# barcode_side_sequencing_primer = Nextera
# barcode_side_sequencing_primer = ACACTCTTTCCCTACACGACGCTCTTCCGATCT : TruSeq and Nextera Illumina sequencing primer on the barcode (i5) side (5' - 3').
common_side_cutsite_remnant = CCG
common_side_sequencing_primer = Truseq
# common_side_sequencing_primer = Nextera
# common_side_sequencing_primer = GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT : TruSeq Illumina sequencing primer on the common (i7) side (5' - 3').
# common_side_sequencing_primer = CGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT : Nextera Illumina sequencing primer on the common (i7) side (5' - 3').
minimum_length = 30
error_rate = 0
output_directory = ./02_trimming
output_file_name_template = {sample_name}_{orientation}{extension}
# Trimming searches and trims the barcode and restriction enzyme cutside remnant at the start of the 5' Forward read, additionally it 3' trims the difference between the maximum barcode length and the current sample's barcode length for each individual read.
# Trimming searches and trims the restriction enzyme cutside remnant at the start of the 5' reverse read.
# Trimming searches for the reverse complement sequence of the common adapter fused to the restriction enzyme cutside remnant and trims these from the 3' of the Forward read.
# Trimming searches for the reverse complement sequence of the barcode adapter fused to the sample-specific barcode and the restriction enzyme cutside remnant and trims these from the 3' of the Reverse read.
# In single-digest GBS, the restriction enzyme cutsite remnant at the common adapter side is the same as on the barcode adapter side.
# Error rate is expressed as fraction of the searched length (range 0.0 - 1.0). 0 means exact match is required. If e.g. for forward reads Truseq adapters (34bp) and MseI (3bp) as a common side RE were used, and a maximum of 1 mismatch is desired, then the error rate should be 0.028. Note that this only allows 1 mismatch if the entire sequence is found. If a smaller portion of the sequence is found, no mismatches will be allowed.
[MaxNFilter]
max_n = 0
output_directory = ./03_max_n_filter
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with N base calls.
[SlidingWindowQualityFilter]
window_size = 2
average_quality = 20
count = 1
output_directory = ./04_sliding_window
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with low quality base calls.
# Translation of default values: For any given read, if any 2 consecutive bases (window_size) have an average Phred quality lower than 20 (average_quality) at least 1 (count) time, then remove the read.
[AverageQualityFilter]
average_quality = 25
output_directory = ./05_average_quality_filter
output_file_name_template = {sample_name}_{run}{extension}
# removes low quality reads.
[RemovePatternFilter.CTGCAG]
pattern = CTGCAG
# first enzyme
output_directory = ./06_remove_chimera_partial_digest
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with intact internal restriction enzyme recognition sites.
[RemovePatternFilter.CCGG]
pattern = CCGG
# second enzyme
output_directory = ./07_remove_chimera_partial_digest
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with intact internal restriction enzyme recognition sites.

[General]
cores = 32
input_directory = /home/User/GBS_preprocessing
sequencing_type = pe
temp_dir = /tmp/
input_file_name_template = {run:24}_R{orientation:1}_001{extension:10}
# Example: 17146FL-13-01-01_S9_L002_R1_001.fastq.bz2 => run = 17146FL-13-01-01_S9_L002; orientation = 1; extension = .fastq.bz2
# Essential information on run, read orientation, and file extension is obtained from the structure of the name of the original fastq file as provided by the service provider.
# {:XX} mark the number of characters in the file name that contain the specified information. One wildcard is allowed, this is invoked by leaving the character length information out. E.g. {run}_R{orientation:1}{extension:10} .
# Fields "orientation" and "extension" are automatically transferred to all new file names created in the next steps.
[CutadaptDemultiplex]
barcodes = /home/User/GBS_preprocessing/barcodes.fasta
error_rate = 0
output_directory = ./01_demultiplex
output_file_name_template = {sample_name}_{orientation}{extension}
anchored_barcodes=True
barcode_side_cutsite_remnant = CTGCA
# the (barcode + barcode_side_cutsite_remnant) sequence is searched per Forward read, to increase specificity of demultiplexing.
# cutsite remnants may also be provided in separate .fasta files where each sample is followed by their corresponding cutsite remnant.
# Error rate is expressed as fraction of the searched length (range 0.0 - 1.0). 0 means exact match is required. If e.g. barcodes used are 5 bp long and the barcode side RE is PstI (5bp), and a maximum of 1 mismatch is desired, then error_rate should be 0.1.
[CutadaptTrimmer]
barcodes = /home/User/GBS_preprocessing/barcodes.fasta
barcode_side_cutsite_remnant = CTGCA
# cutsite remnants may also be provided in separate .fasta files where each sample is followed by their corresponding cutsite remnant.
barcode_side_sequencing_primer = TruSeq
# barcode_side_sequencing_primer = Nextera
# barcode_side_sequencing_primer = ACACTCTTTCCCTACACGACGCTCTTCCGATCT : TruSeq and Nextera Illumina sequencing primer on the barcode (i5) side (5' - 3').
common_side_cutsite_remnant = CCG
common_side_sequencing_primer = Truseq
# common_side_sequencing_primer = Nextera
# common_side_sequencing_primer = GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT : TruSeq Illumina sequencing primer on the common (i7) side (5' - 3').
# common_side_sequencing_primer = CGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT : Nextera Illumina sequencing primer on the common (i7) side (5' - 3').
minimum_length = 30
error_rate = 0
output_directory = ./02_trimming
output_file_name_template = {sample_name}_{orientation}{extension}
# Trimming searches and trims the barcode and restriction enzyme cutside remnant at the start of the 5' Forward read, additionally it 3' trims the difference between the maximum barcode length and the current sample's barcode length for each individual read.
# Trimming searches and trims the restriction enzyme cutside remnant at the start of the 5' reverse read.
# Trimming searches for the reverse complement sequence of the common adapter fused to the restriction enzyme cutside remnant and trims these from the 3' of the Forward read.
# Trimming searches for the reverse complement sequence of the barcode adapter fused to the sample-specific barcode and the restriction enzyme cutside remnant and trims these from the 3' of the Reverse read.
# In single-digest GBS, the restriction enzyme cutsite remnant at the common adapter side is the same as on the barcode adapter side.
# Error rate is expressed as fraction of the searched length (range 0.0 - 1.0). 0 means exact match is required. If e.g. for forward reads Truseq adapters (34bp) and MseI (3bp) as a common side RE were used, and a maximum of 1 mismatch is desired, then the error rate should be 0.028. Note that this only allows 1 mismatch if the entire sequence is found. If a smaller portion of the sequence is found, no mismatches will be allowed.
[Pear]
minimum_length = 40
minimum_overlap = 10
output_directory = ./03_merging
output_file_name_template = {sample_name}_{run}.assembled{extension}
# merges Forward and Reverse reads by sequence overlap.
[MaxNFilter]
max_n = 0
output_directory = ./04_max_n_filter
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with N base calls.
[SlidingWindowQualityFilter]
window_size = 2
average_quality = 20
count = 1
output_directory = ./05_sliding_window
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with low quality base calls.
# Translation of default values: For any given read, if any 2 consecutive bases (window_size) have an average Phred quality lower than 20 (average_quality) at least 1 (count) time, then remove the read.
[AverageQualityFilter]
average_quality = 25
output_directory = ./06_average_quality_filter
output_file_name_template = {sample_name}_{run}{extension}
# removes low quality reads.
[RemovePatternFilter.CTGCAG]
pattern = CTGCAG
# first enzyme
output_directory = ./07_remove_chimera_partial_digest
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with intact internal restriction enzyme recognition sites.
[RemovePatternFilter.CCGG]
pattern = CCGG
# second enzyme
output_directory = ./08_remove_chimera_partial_digest
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with intact internal restriction enzyme recognition sites.

[General]
cores = 32
input_directory = /home/User/GBS_preprocessing
sequencing_type = pe
temp_dir = /tmp/
input_file_name_template = {run:24}_R{orientation:1}_001{extension:10}
# Example: 17146FL-13-01-01_S9_L002_R1_001.fastq.bz2 => run = 17146FL-13-01-01_S9_L002; orientation = 1; extension = .fastq.bz2
# Essential information on run, read orientation, and file extension is obtained from the structure of the name of the original fastq file as provided by the service provider.
# {:XX} mark the number of characters in the file name that contain the specified information. One wildcard is allowed, this is invoked by leaving the character length information out. E.g. {run}_R{orientation:1}{extension:10} .
# Fields "orientation" and "extension" are automatically transferred to all new file names created in the next steps.
[CutadaptDemultiplex]
barcodes = /home/User/GBS_preprocessing/barcodes.fasta
error_rate = 0
output_directory = ./01_demultiplex
output_file_name_template = {sample_name}_{orientation}{extension}
anchored_barcodes=True
barcode_side_cutsite_remnant = CTGCA
# the (barcode + barcode_side_cutsite_remnant) sequence is searched per read, to increase specificity of demultiplexing.
# cutsite remnants may also be provided in separate .fasta files where each sample is followed by their corresponding cutsite remnant.
# Error rate is expressed as fraction of the searched length (range 0.0 - 1.0). 0 means exact match is required. If e.g. barcodes used are 5 bp long and the barcode side RE is PstI (5bp), and a maximum of 1 mismatch is desired, then error_rate should be 0.1.
[CutadaptTrimmer]
barcodes = /home/User/GBS_preprocessing/barcodes.fasta
barcode_side_cutsite_remnant = CTGCA
# cutsite remnants may also be provided in separate .fasta files where each sample is followed by their corresponding cutsite remnant.
barcode_side_sequencing_primer = TruSeq
# barcode_side_sequencing_primer = Nextera
# barcode_side_sequencing_primer = ACACTCTTTCCCTACACGACGCTCTTCCGATCT : TruSeq and Nextera Illumina sequencing primer on the barcode (i5) side (5' - 3').
common_side_cutsite_remnant = CCG
common_side_sequencing_primer = Truseq
# common_side_sequencing_primer = Nextera
# common_side_sequencing_primer = GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT : TruSeq Illumina sequencing primer on the common (i7) side (5' - 3').
# common_side_sequencing_primer = CGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT : Nextera Illumina sequencing primer on the common (i7) side (5' - 3').
spacer = AGTC
minimum_length = 30
error_rate = 0
output_directory = ./02_trimming
output_file_name_template = {sample_name}_{orientation}{extension}
# Trimming searches and trims the barcode and restriction enzyme cutside remnant at the start of the 5' Forward read, additionally it 3' trims the difference between the maximum barcode length and the current sample's barcode length for each individual read.
# Trimming searches and trims the spacer and restriction enzyme cutside remnant at the start of the 5' Reverse read, additionally it 3' trims the difference between the maximum spacer length and the current read's spacer length for each individual read.
# Trimming searches for the reverse complement sequence of the common adapter fused to the restriction enzyme cutside remnant and trims these from the 3' of the Forward read.
# Trimming searches for the reverse complement sequence of the barcode adapter fused to the sample-specific barcode and the restriction enzyme cutside remnant and trims these from the 3' of the Reverse read.
# In single-digest GBS, the restriction enzyme cutsite remnant at the common adapter side is the same as on the barcode adapter side.
# Error rate is expressed as fraction of the searched length (range 0.0 - 1.0). 0 means exact match is required. If e.g. for forward reads Truseq adapters (34bp) and MseI (3bp) as a common side RE were used, and a maximum of 1 mismatch is desired, then the error rate should be 0.028. Note that this only allows 1 mismatch if the entire sequence is found. If a smaller portion of the sequence is found, no mismatches will be allowed.
[MaxNFilter]
max_n = 0
output_directory = ./03_max_n_filter
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with N base calls.
[SlidingWindowQualityFilter]
window_size = 2
average_quality = 20
count = 1
output_directory = ./04_sliding_window
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with low quality base calls.
# Translation of default values: For any given read, if any 2 consecutive bases (window_size) have an average Phred quality lower than 20 (average_quality) at least 1 (count) time, then remove the read.
[AverageQualityFilter]
average_quality = 25
output_directory = ./05_average_quality_filter
output_file_name_template = {sample_name}_{run}{extension}
# removes low quality reads.
[RemovePatternFilter.CTGCAG]
pattern = CTGCAG
# first enzyme
output_directory = ./06_remove_chimera_partial_digest
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with intact internal restriction enzyme recognition sites.
[RemovePatternFilter.CCGG]
pattern = CCGG
# second enzyme
output_directory = ./07_remove_chimera_partial_digest
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with intact internal restriction enzyme recognition sites.

[General]
cores = 32
input_directory = /home/User/GBS_preprocessing
sequencing_type = pe
temp_dir = /tmp/
input_file_name_template = {run:24}_R{orientation:1}_001{extension:10}
# Example: 17146FL-13-01-01_S9_L002_R1_001.fastq.bz2 => run = 17146FL-13-01-01_S9_L002; orientation = 1; extension = .fastq.bz2
# Essential information on run, read orientation, and file extension is obtained from the structure of the name of the original fastq file as provided by the service provider.
# {:XX} mark the number of characters in the file name that contain the specified information. One wildcard is allowed, this is invoked by leaving the character length information out. E.g. {run}_R{orientation:1}{extension:10} .
# Fields "orientation" and "extension" are automatically transferred to all new file names created in the next steps.
[CutadaptDemultiplex]
barcodes = /home/User/GBS_preprocessing/barcodes.fasta
error_rate = 0
output_directory = ./01_demultiplex
output_file_name_template = {sample_name}_{orientation}{extension}
anchored_barcodes=True
barcode_side_cutsite_remnant = CTGCA
# the (barcode + barcode_side_cutsite_remnant) sequence is searched per Forward read, to increase specificity of demultiplexing.
# cutsite remnants may also be provided in separate .fasta files where each sample is followed by their corresponding cutsite remnant.
# Error rate is expressed as fraction of the searched length (range 0.0 - 1.0). 0 means exact match is required. If e.g. barcodes used are 5 bp long and the barcode side RE is PstI (5bp), and a maximum of 1 mismatch is desired, then error_rate should be 0.1.
[CutadaptTrimmer]
barcodes = /home/User/GBS_preprocessing/barcodes.fasta
barcode_side_cutsite_remnant = CTGCA
# cutsite remnants may also be provided in separate .fasta files where each sample is followed by their corresponding cutsite remnant.
barcode_side_sequencing_primer = TruSeq
# barcode_side_sequencing_primer = Nextera
# barcode_side_sequencing_primer = ACACTCTTTCCCTACACGACGCTCTTCCGATCT : TruSeq and Nextera Illumina sequencing primer on the barcode (i5) side (5' - 3').
common_side_cutsite_remnant = CCG
common_side_sequencing_primer = Truseq
# common_side_sequencing_primer = Nextera
# common_side_sequencing_primer = GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT : TruSeq Illumina sequencing primer on the common (i7) side (5' - 3').
# common_side_sequencing_primer = CGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT : Nextera Illumina sequencing primer on the common (i7) side (5' - 3').
spacer = AGTC
minimum_length = 30
error_rate = 0
output_directory = ./02_trimming
output_file_name_template = {sample_name}_{orientation}{extension}
# Trimming searches and trims the barcode and restriction enzyme cutside remnant at the start of the 5' Forward read, additionally it 3' trims the difference between the maximum barcode length and the current sample's barcode length for each individual read.
# Trimming searches and trims the spacer and restriction enzyme cutside remnant at the start of the 5' Reverse read, additionally it 3' trims the difference between the maximum spacer length and the current read's spacer length for each individual read.
# Trimming searches for the reverse complement sequence of the common adapter fused to the restriction enzyme cutside remnant and trims these from the 3' of the Forward read.
# Trimming searches for the reverse complement sequence of the barcode adapter fused to the sample-specific barcode and the restriction enzyme cutside remnant and trims these from the 3' of the Reverse read.
# In single-digest GBS, the restriction enzyme cutsite remnant at the common adapter side is the same as on the barcode adapter side.
# Error rate is expressed as fraction of the searched length (range 0.0 - 1.0). 0 means exact match is required. If e.g. for forward reads Truseq adapters (34bp) and MseI (3bp) as a common side RE were used, and a maximum of 1 mismatch is desired, then the error rate should be 0.028. Note that this only allows 1 mismatch if the entire sequence is found. If a smaller portion of the sequence is found, no mismatches will be allowed.
[Pear]
minimum_length = 40
minimum_overlap = 10
output_directory = ./03_merging
output_file_name_template = {sample_name}_{run}.assembled{extension}
# merges Forward and Reverse reads by sequence overlap.
[MaxNFilter]
max_n = 0
output_directory = ./04_max_n_filter
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with N base calls.
[SlidingWindowQualityFilter]
window_size = 2
average_quality = 20
count = 1
output_directory = ./05_sliding_window
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with low quality base calls.
# Translation of default values: For any given read, if any 2 consecutive bases (window_size) have an average Phred quality lower than 20 (average_quality) at least 1 (count) time, then remove the read.
[AverageQualityFilter]
average_quality = 25
output_directory = ./06_average_quality_filter
output_file_name_template = {sample_name}_{run}{extension}
# removes low quality reads.
[RemovePatternFilter.CTGCAG]
pattern = CTGCAG
# first enzyme
output_directory = ./07_remove_chimera_partial_digest
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with intact internal restriction enzyme recognition sites.
[RemovePatternFilter.CCGG]
pattern = CCGG
# second enzyme
output_directory = ./08_remove_chimera_partial_digest
output_file_name_template = {sample_name}_{run}{extension}
# removes reads with intact internal restriction enzyme recognition sites.
Starting after Merging
[General] cores = 32 input_directory = /home/User/GBS_preprocessing/03_merging sequencing_type = se # After merging, only a single FASTQ file remains per sample. This file is then essentially equal to single-end FASTQ files and therefore the sequencing type should be se. temp_dir = /tmp/ input_file_name_template = {sample_name:35}.assembled{extension:10} # Example: 006_015_170516_001_0251_069_01_1081.assembled.fastq.bz2 => sample_name = 17146FL-13-01-01_S9_L002; extension = .fastq.bz2 # Essential information on run, read orientation, and file extension is obtained from the structure of the name of the original fastq file as provided by the service provider. # {:XX} mark the number of characters in the file name that contain the specified information. One wildcard is allowed, this is invoked by leaving the character length information out. E.g. {run}_R{orientation:1}{extension:10} . # Fields "orientation" and "extension" are automatically transferred to all new file names created in the next steps. [MaxNFilter] max_n = 0 output_directory = ./05_max_n_filter output_file_name_template = {sample_name}{extension} # removes reads with N base calls. [SlidingWindowQualityFilter] window_size = 2 average_quality = 20 count = 1 output_directory = ./06_sliding_window output_file_name_template = {sample_name}{extension} # removes reads with low quality base calls. # Translation of default values: For any given read, if any 2 consecutive bases (window_size) have an average Phred quality lower than 20 (average_quality) at least 1 (count) time, then remove the read. [AverageQualityFilter] average_quality = 25 output_directory = ./07_average_quality_filter output_file_name_template = {sample_name}{extension} # removes low quality reads. [RemovePatternFilter.CTGCAG] pattern = CTGCAG # first enzyme output_directory = ./08_remove_chimera_partial_digest output_file_name_template = {sample_name}{extension} # removes reads with intact internal restriction enzyme recognition sites. [RemovePatternFilter.CCGG] pattern = CCGG # second enzyme output_directory = ./09_remove_chimera_partial_digest output_file_name_template = {sample_name}{extension} # removes reads with intact internal restriction enzyme recognition sites.