GBS data preprocessing: How it works
Read preprocessing steps
NGS read preprocessing requires specific steps for single-digest or double-digest GBS, in combination with single-end or paired-end sequenced reads. Paired-end reads may be mapped separately or merged with (PEAR) before mapping.
This page provides a visual overview of the GBprocesS workflow, including the library preparation steps prior to GBprocesS:
Library preparation and sequencing.
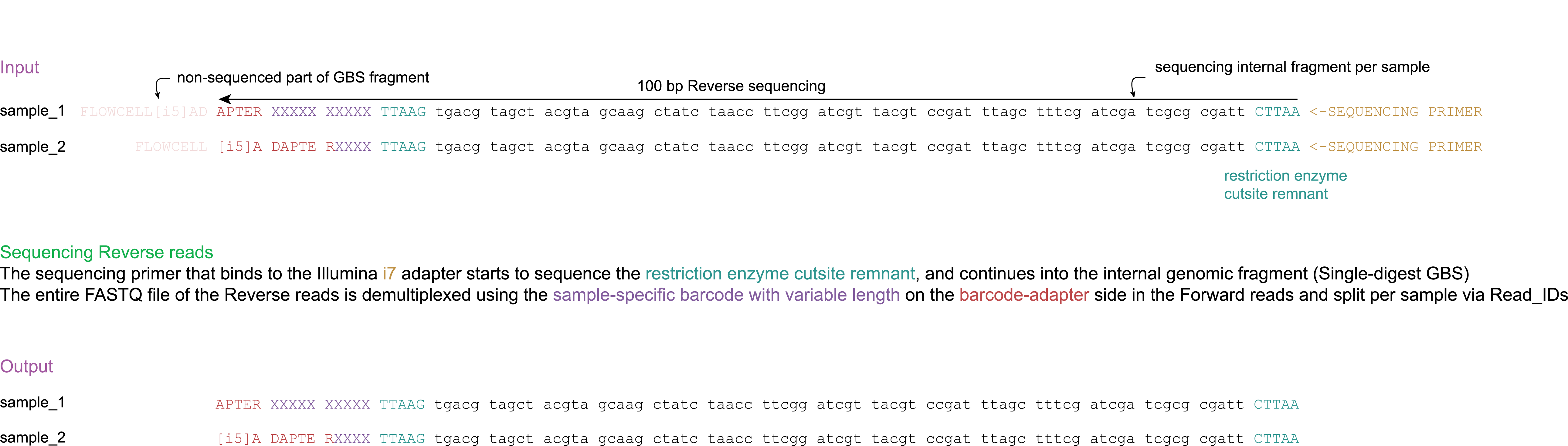
Demultiplexing (Cutadapt).
Trimming: Trimming barcodes (or spacers) and restriction enzyme cutsite remnants at the 5’ end of the reads (while compensating for variable length barcodes and spacers by trimming at the 3’ end of forward reads) and trimming of restriction enzyme cutsite remnants, barcodes (or spacers), and adapter sequences at the 3’ end of the reads. (Cutadapt).
Merging of forward and reverse reads.
Removal of reads with low quality base-calling (Python).
Removal of reads with internal restriction sites (Python).
What goes on, must come off
Barcodes and adapter sequences must be removed because these do not occur in the reference genome and would lead to errors in read mapping and/or identification of polymorphisms that do not exist. Restriction site remnants are removed as these would create positional overlaps of neighboring stacks derived from independent PCR-amplified GBS fragments. We recommend to remove reads with overall low quality and with internal restriction sites. Clipping of reads with a sliding window that clips reads when base calling quality within a window drops below a specified threshold is not recommended. This will create reads with variable length, and thus mappings of variable length, which reflect technical artefacts and not biologically meaningfull sequence diversity.
0. Library preparation & Sequencing
The tabs below contain a visual overview of the regular GBS library preparation steps. The images are examples of single-digest GBS library preparation and paired-end sequencing, the tab Ligation includes several different cases of adapters.


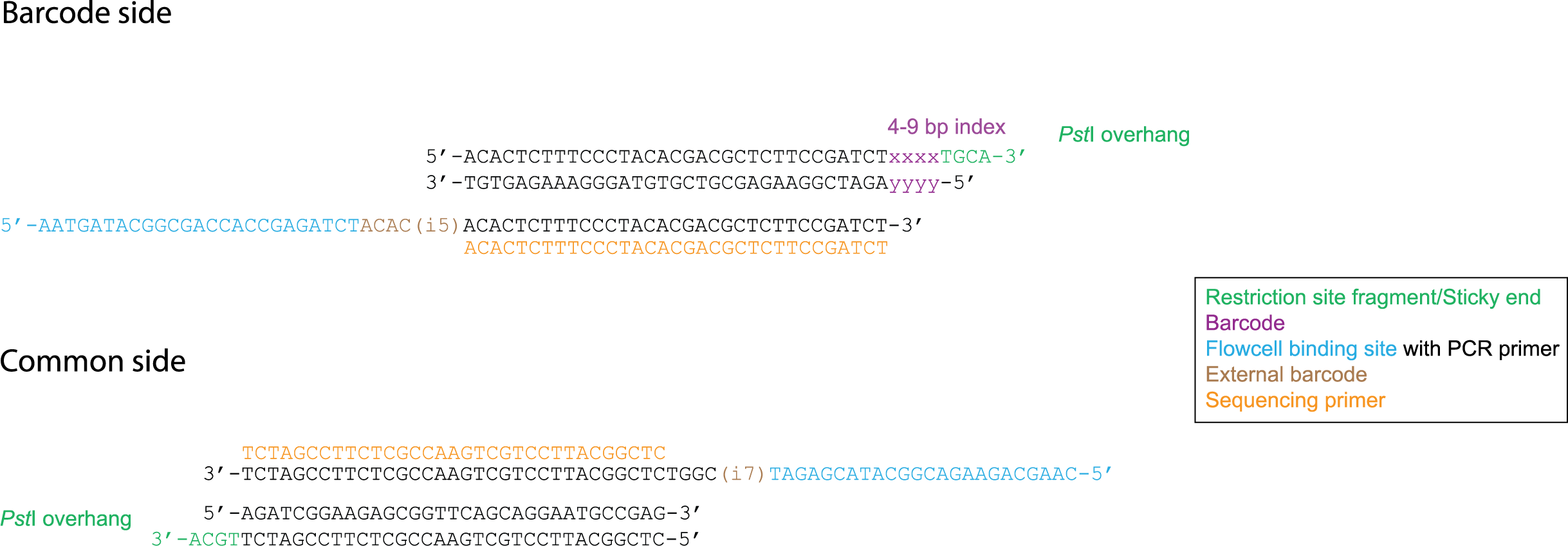
Poland et al. (2012) developped the concept of reverse Y-shaped adapters for GBS. Y-shaped adapters are not targettable by primers due to their single strandedness, therefore only forward strand transcripts can be amplified during PCR. Subsequently these forward primers generate the complementary sequence of the reverse primer, after which reverse primers are able to start sequence generation. Reverse Y-shaped adapters are mainly used for frequent cutting enzymes in order to reduce PCR amplification competition with rare-cutting enzyme fragments. Additionally this method allows for the combination of both PCR cycles into 1 cycle. If a rare cutting restriction site is represented by A and a frequenct cutting restriction site is represented by a B, then the PCR product will only consist of ds AB and ss A.
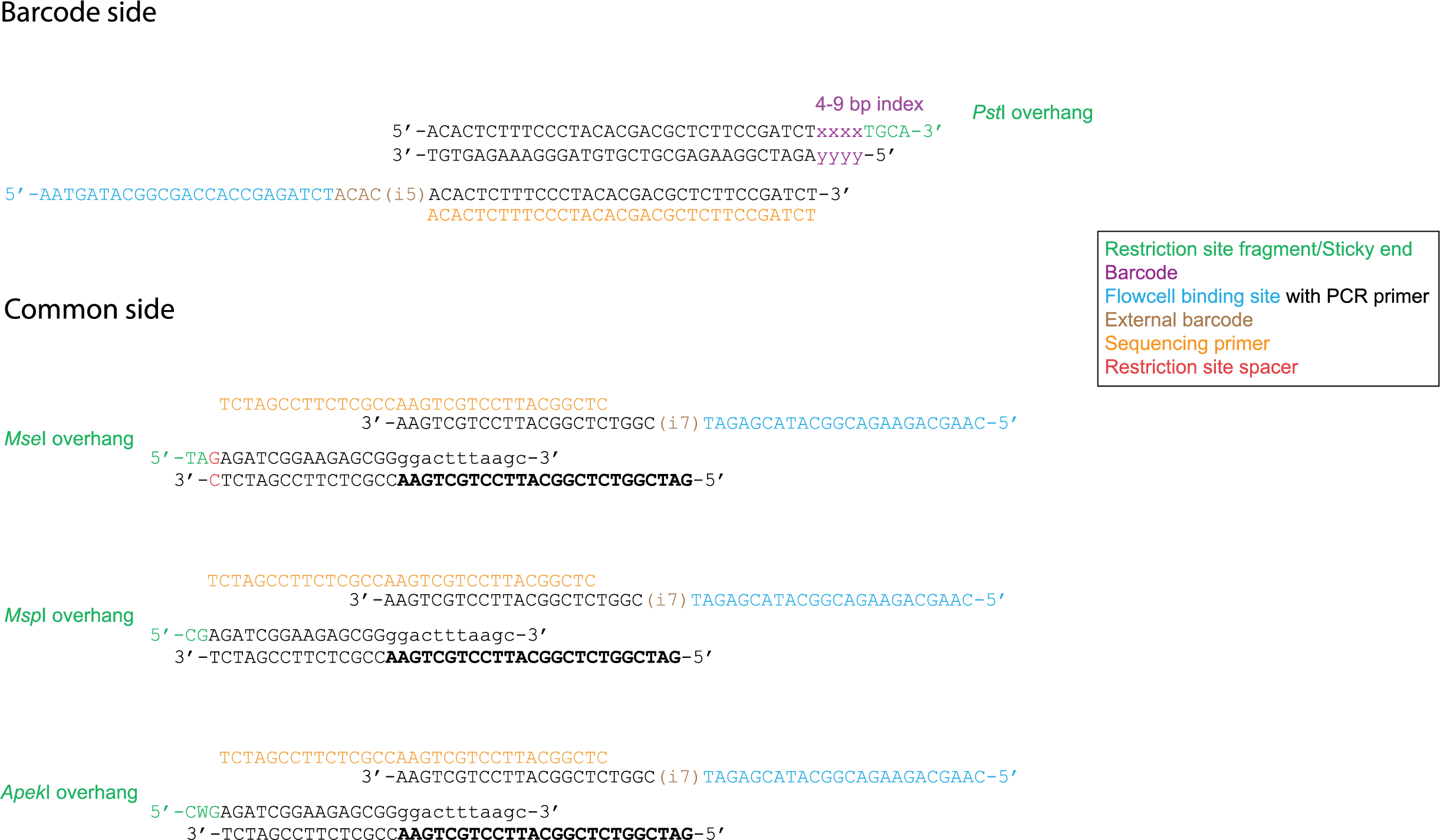
For an explanation about the restriction site spacer in Mse I, see tab “Special adapter cases”.
Illumina sequencing determines base identities at the hand of spectroscopy, however when there is little variation between basecalls, the device can malfuction. It is therefore necessary to apply some kind of phazing (length polymorphisms) within adapters. Typically, this length variability is introduced at the barcode adapter side at the hand of variable length internal and external barcodes. However, sometimes a variable spacer is introduced in the common adapter in order to increase phazing. These kind of data are processed using the spacer option in the Trimming operation.
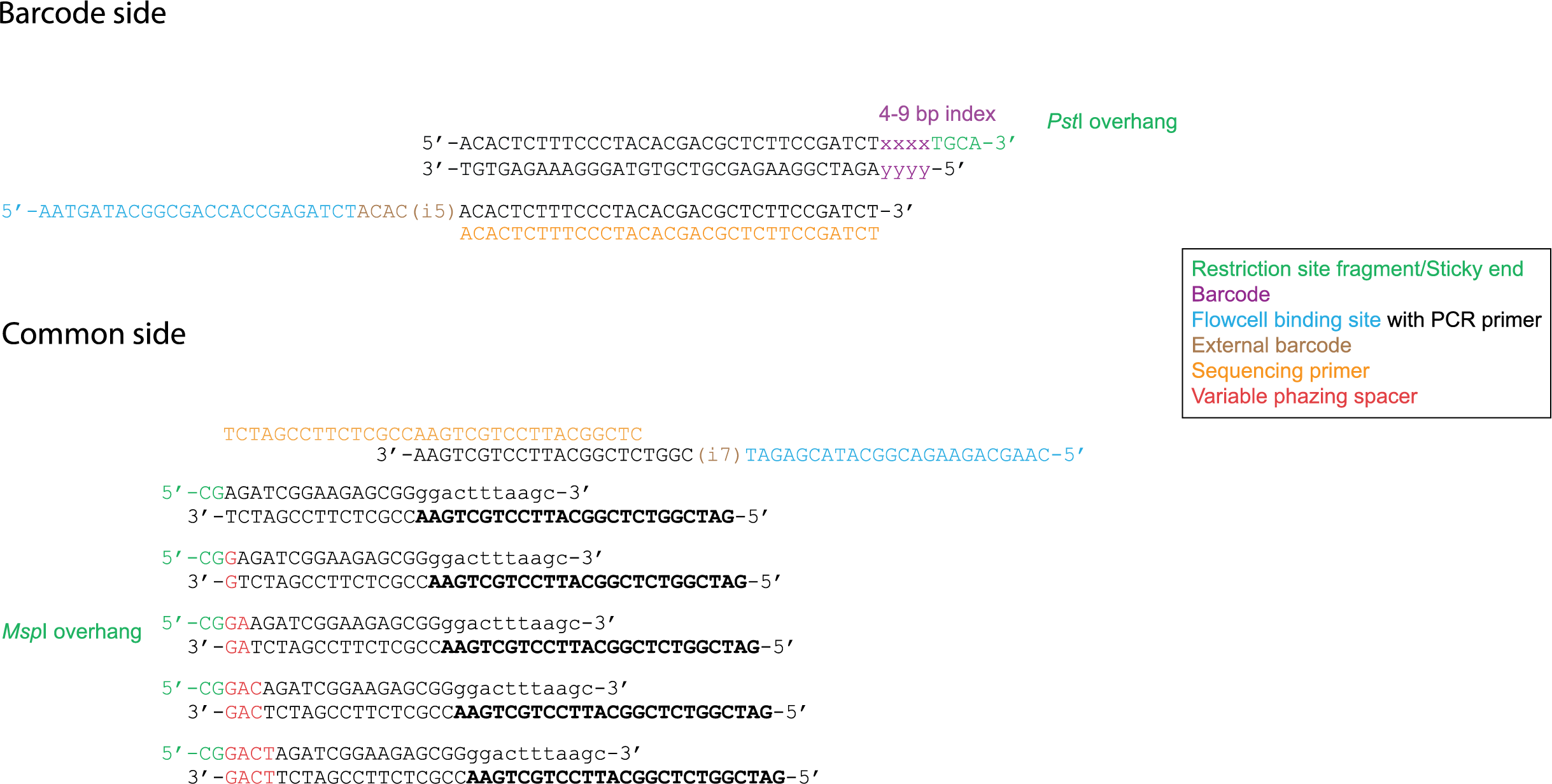
By introducing a G directly after the sticky end, the Msp I restriction site is restored. Therefore it is not possible to add restriction enzymes and adapters in the same library preparation step.
The first adapters released by Illumina were called Nextera. Later TruSeq was released; these adapters are exactly the same on the barcode side but different on the common side.

Sometimes restriction enzymes and adapters are added to the extracted DNA at the same time in order to simplify the process. However in certain cases when adapters are added a new restriction site is created (e.g. in Mse I), effectively destroying the adapter. Therefore a 1 bp spacer is added in order to counteract this effect. This 1bp spacer should be included in the config file as part of the restriction site fragment.
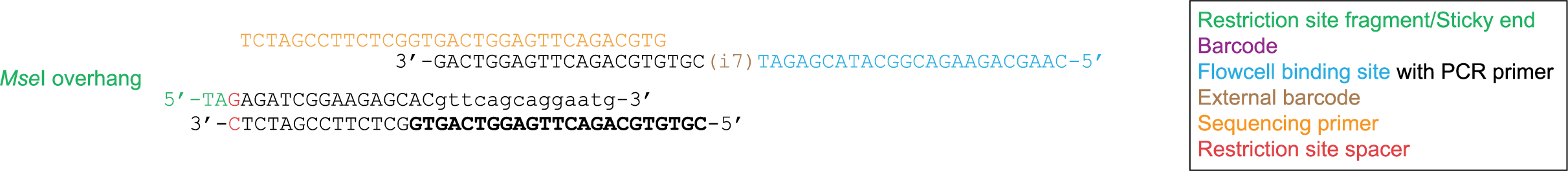

Forward read 75 bp

Forward read 100 bp

Reverse read 75 bp

Reverse read 100 bp
1. Demultiplexing
Operation: CutadaptDemultiplex

2. Trimming
Operation: Trimming




3. Merging
Operation: Pear


Quality filtering
Quality filtering removes entire reads based on Phred basecall quality scores or Illumina N-base calls. Additional information on how Phred Q scores relate to base call accuracy can be found e.g. here.
Sliding Window Quality Filter
Operation: SlidingWindowQualityFilter
The ASCII quality score line of the .fastq records are interpreted using the Illumina 1.8+ Phred+33 encoding. At the hand of a Sliding Window with custom window size (5 below) and set step size 1 .fastq reads are divided into smaller segments. Within these segments (windows), window average quality scores are calculated. If a window average quality score falls below a custom set quality threshold (25 below and depicted by the red boxes), +1 is added to the bad window counts. At the end of the evaluation of the entire read, the bad window count is compared with the count threshold (1 below). If the bad window count exceeds the threshold, the read is discarded.

Average Quality Filter
Operation: AverageQualityFilter
The ASCII quality score line of the .fastq records are interpreted using the Illumina 1.8+ Phred+33 encoding. An average of the resulting integers is compared to a set threshold. Reads with an average quality score below this threshold are discarded.
Max N Filter
Operation: MaxNFilter
Read filtering based on maximum amount of Illumina N-base calls (any base). Removes reads with amounts of N above set threshold
Read sequence filtering
Operation: RemovePatternFilter
Internal sequence filtering removes entire reads that contain a given sequence by exact matching. Most commonly used to remove reads with intact restriction sites, but can be used for other purposes.
Read length filtering
Operation: LengthFilter
Read length filtering removes entire reads that are shorter than a minimal length.
Why merge?
If paired-end data is obtained, two stategies can be applied. The first strategy maps both reads separately. The benefits are that no reads are lost due to not reaching the minimum_overlap length required for merging reads with PEAR. The drawbacks are that Stacks of forward reads and Stacks of reverse reads partially overlap (carry redundant information), can inflate read depth and smaller 'haplotypes’ are considered than when reads are merged.
The second strategy first merges the forward and reverse read per GBS-amplified fragment, thus creating a longer single read. The benefit is that because the single resulting read may span more neighboring SNPs, thus extending the potential length of local haplotypes, removing the redundancy observed in separately mapped reads, and that read depth is maximal per locus. The most important point of attention is that a particular minimal length of the overlap between the forward and reverse reads must be chosen during the merging process. Picking a minimum merging overlap length is a trade-off between sacrificing long loci, and removing false positive overlaps, it is recommended to at least use a merging overlap of 10 to remove most of the false positive merged reads, see also Figure 6 in Magoč T. & Salzberg S., 2011.